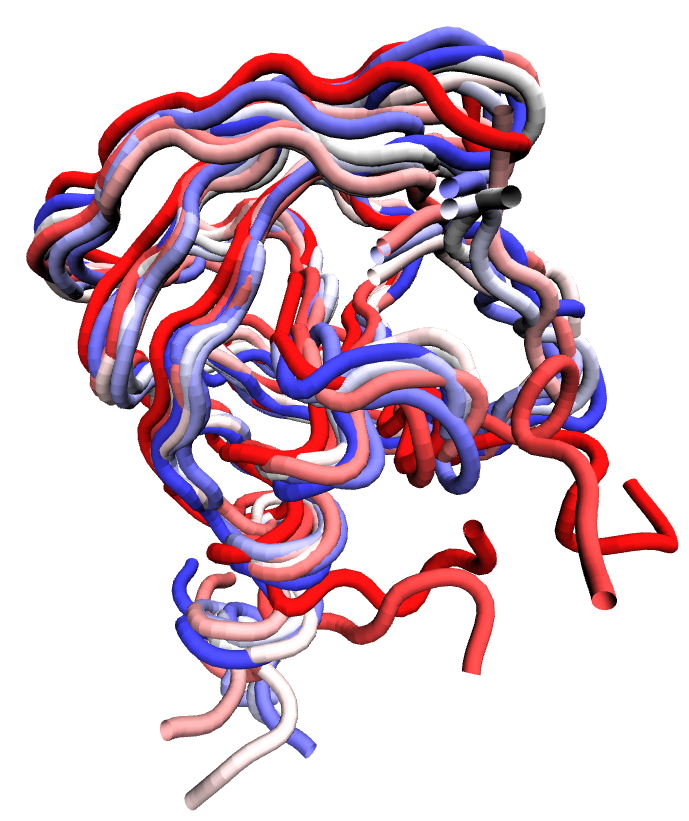
Conformational ensemble of HIV-1 protease (PDB ID: 3TTP) backbone atoms harvested from a one microsecond equilibrium molecular dynamics simulation run at constant temperature and pressure. Each backbone is colored based on timestep in the ensemble.
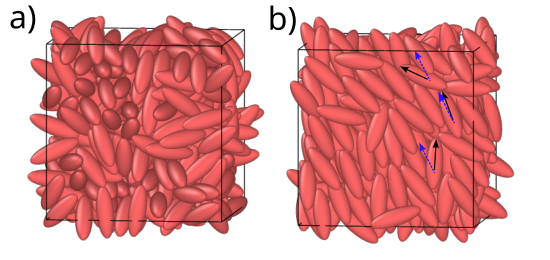
Isotropic (left, a) and nematic (right, b) phases of a melt of Gay-Berne ellipsoids. In b), the local orientations of each ellipsoid relative to the global orientation vector
(blue, dashed arrow) are indicated with the black arrows for a few sample ellipsoids.